
ISO13485 医疗器械质量管理体系
发布时间:
ISO13485质量管理体系认证
一、服务企业:
(1)第一类医疗器械生产/销售企业;
(2)第二三类医疗器械生产/销售企业;
(3)欲申请医疗器械注册和生产或经营的企业。
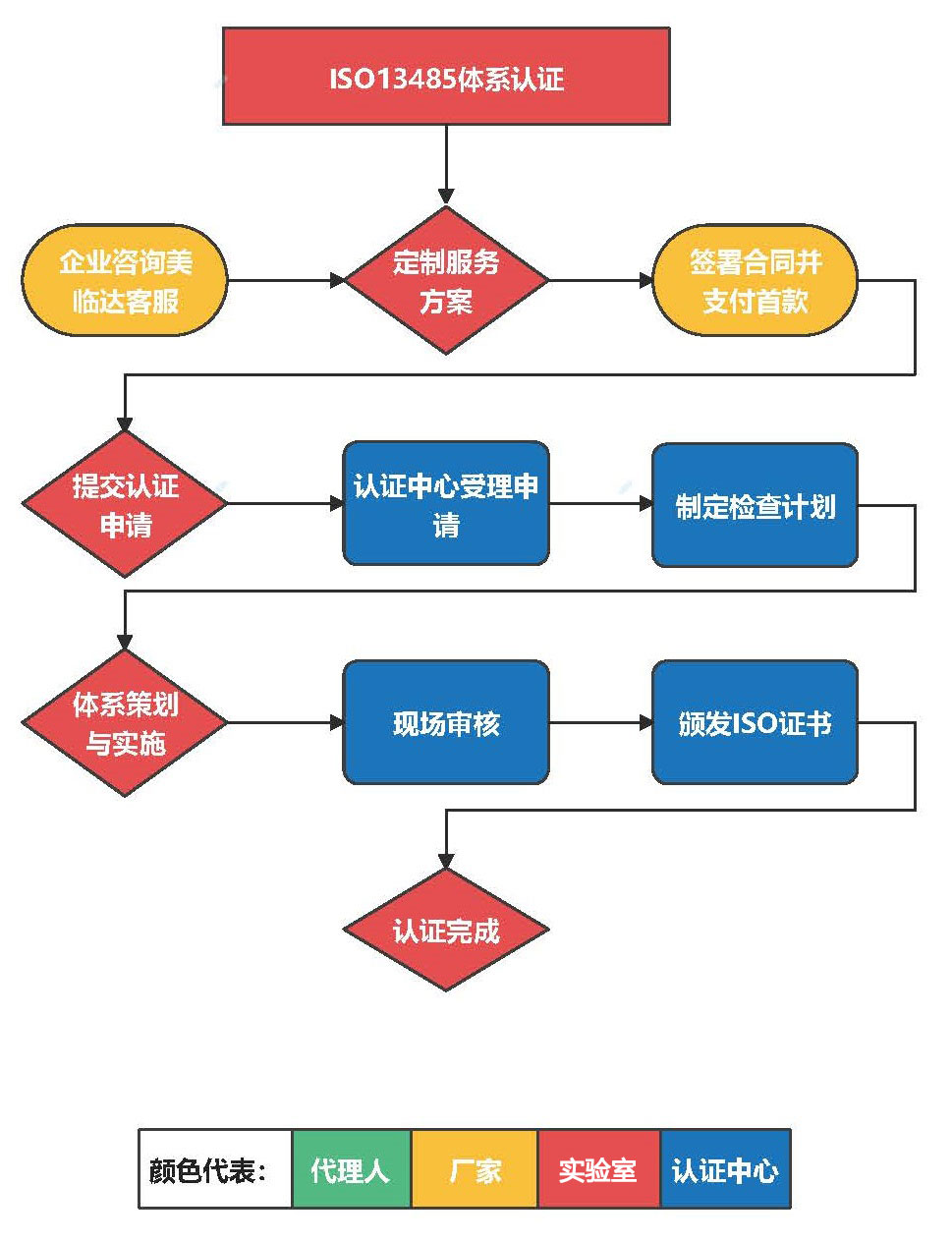
二、服务流程:
A)初次认证的流程
1- 企业提交《认证申请表》,连同认证中心要求的材料一同报给认证中心;
2- 认证中心受理认证申请;
3- 成立检查组,制定检查计划并通报给企业确认;
4- 检查小组现场审核生产环境、生产能力、生产记录等,撰写评价报告,提交审查。
5- 认证中心收到技术委审查意见后,汇总审查意见;
6- 认证中心向合格的企业颁发《ISO13485认证证书》中英文双语证书;
7- 组织公告和宣传,认证完成。
B)年度监督检查
1- 认证中心成立检查组,制定检查计划并通报给企业确认;
2- 检查小组现场审核生产环境、生产能力、生产记录等,撰写评价报告,提交审查。
3- 认证中心收到技术委审查批准意见后,汇总审查意见;
4- 年度监督检查每年一次。
C)复评认证
3年到期的企业,应重新填写《ISO13485认证申请表》,连同有关材料报认证中心。其余认证程序同初次认证。
三、体系认证准备:
准备1:
-申请组织应持有法人营业执照或证明其法律地位的文件。
准备2:
-已取得生产许可证或其它资质证明(国家或部门法规有要求时);
准备3:
-申请认证的质量管理体系覆盖的产品应符合有关国家标准、行业标准或注册产品标准(企业标准),产品定型且成批生产。
准备4:
-申请组织应建立符合拟申请认证标准的管理体系、对医疗器械生产、经营企业还应符合YY/T 0287标准的要求,生产三类医疗器械的企业,质量管理体系运行时间不少于6个月, 生产和经营其它产品的企业,质量管理体系运行时间不少于3个月。并至少进行过一次全面内部审核及一次管理评审。 质量体系的相关文件可委托第三方机构进行撰写整理。
准备5:
-在提出认证申请前的一年内,申请组织的产品无重大顾客投诉及质量事故。
准备6:
-ISO13485质量准备文件:三级文件
第一级文件:质量手册
第二级文件:程序文件
第三极文件:记录文件、表格、作业指导书、技术标准、图纸、操作规程、部门制度等

四、服务依据:
CFDA关于发布医疗器械生产质量管理规范的公告(2014年第64号)
食药监械监〔2015〕218号附件1-医疗器械生产质量管理规范现场检查指导原则
食药监械监〔2015〕218号附件4-医疗器械生产质量管理规范体外诊断试剂现场检查指导原则
YY/T 0287-2017 idt ISO 13485:2016 《医疗器械 质量管理体系 用于法规的要求》
YY/T 0316-2016 idt ISO 14971:2007 《医疗器械 风险管理对医疗器械的应用》

五、注意事项:
认证分为初次认证、年度监督检查和复评认证。为了保证体系的有效运行,在企业初次认证成功后,每年由审核机构进行年度审查。ISO13485认证有效期为3年,有效期满前需进行复评认证。
其次:企业在认证之前需取得相关的资质证书,具体如下:
|
从业项目 |
须取得的资质 |
|
一类医疗器械生产 |
《第一类医疗器械产品备案凭证》+《第一类医疗器械生产备案凭证》 |
|
二三类医疗器械生产 |
《医疗器械注册证》+《医疗器械生产许可证》 |
|
二类医疗器械销售 |
《第二类医疗器械经营备案凭证》 |
|
三类医疗器械销售 |
《第三类医疗器械经营许可证》 |
取得以上资质后3-6个月后即可申请ISO13485认证。(对于生产三类医疗器械产品,体系运行时间至少6个月,其他产品的管理体系至少运行3个月)。
六、咨询方式:
无